- Página de Entrada
- Síndromes Hereditárias
Síndromes Hereditárias
share
O que são?
O cancro é uma doença multifatorial, que tem origem em "erros”/mutações que permanecem no ADN das células. Na maioria das vezes o cancro é esporádico, isto é, resulta do envelhecimento e da exposição a agentes que podem lesar o ADN (tabaco, álcool, obesidade...). Portanto, em cerca de 90% dos casos, o cancro não é herdado, nem se transmite à descendência.
Em cerca de 10% de todos os casos de cancro, o "erro”/mutação do ADN que poderá causar a doença, está presente em todas as células do indivíduo, inclusive nas suas células sexuais ou germinativas (óvulos e espermatozóides) e pode ser transmitido à descendência. Pelo que, 10% de todos os cancros surge no contexto de síndrome de predisposição familiar para o cancro (herança da predisposição para, não do cancro).
As síndromes de predisposição familiar para o cancro, são doenças que aumentam a predisposição para o desenvolvimento de cancro ao longo da vida. Estas resultam de mutações germinativas em genes importantes para o desenvolvimento de tumores (por exemplo: genes supressores de tumor). Na maioria dos casos, os genes mutados são transmitidos de pais para filhos, causando cancro em todas as gerações (herança autossómica dominante). Em teoria, um portador de mutação tem probabilidade de transmitir a mutação a 50% dos seus descendentes. Pode acontecer que pessoas portadoras do gene mutado, nunca venham a desenvolver cancro, mesmo em idade avançada. Chama-se a isto penetrância incompleta. No entanto, a herança do gene mutado traz consigo uma alta probabilidade de a pessoa vir a desenvolver cancro. Esta probabilidade varia de síndrome para síndrome.
Principais Síndromes Hereditários de Cancro:
- Síndrome Hereditário de Cancro da Mama/Ovário
-
O que é a HBOC?A Síndrome Hereditário de Cancro da Mama e Ovário (HBOC) é responsável por cerca de 5 a 10% dos casos diagnosticados de neoplasia da mama, cerca de 15% dos casos de cancro do ovário e 3% dos casos de cancro da próstata e pâncreas. Está associado a mutações em vários genes, sendo que na maioria dos casos se associa com mutações germinativas nos genes BRCA1 e BRCA2.O que causa HBOC?A maior parte do HBOC está associado a uma alteração em 2 genes BRCA1 e BRCA2, conhecidos como genes supressores tumorais, cuja função é regular a divisão das células e a morte celular programada. As proteínas codificadas por estes genes conseguem reparar erros genéticos, que resultam da divisão celular. Se estas proteínas estiverem deficientes, os erros não são reparados e podem originar células anormais (cancro).
Como se manifesta?Os portadores de alterações patogénicas nos genes BRCA1 ou BRCA2 têm um risco aumentado de poderem desenvolver cancro da mama ou do ovário o longo da sua vida:O risco de desenvolver cancro da mama numa mulher com mutação patogénica no gene BRCA1 é de cerca de 57%, enquanto se o gene afetado for o BRCA2 esse risco será um pouco inferior, cerca de 50%.Relativamente ao risco de desenvolver cancro do ovário:• uma mulher com mutação patogénica BRCA1 tem 40% de risco de desenvolver cancro do ovário e com mutação patogénica BRCA2 tem cerca de 20%.Também há um aumento do risco para o aparecimento de outros cancros, sobretudo cancro do pâncreas, cancro do estômago, cancro da pele (melanoma maligno) e nos homens risco aumentado de cancro da próstata.Probabilidade de Cancro ao longo da vida Cancro da Mama Cancro do Ovário Cancro do Pâncreas População Geral 13% 1-2% 1-2% BRCA1 57% 40% <5% BRCA2 49%20% 5-10%
Contudo, nem todas as pessoas com variantes patogénicas germinativas BRCA1 ou BRCA2 vêm a ter cancro.Outros síndromes hereditários mais raros, associados aos genes TP53 (Sindrome de Li-Fraumeni) e PTEN (Sindrome de Cowden) também se associam a um elevado risco de cancro da mama. A Síndrome de Li-Fraumeni será abordada em secção própria.Outros genes com risco aumentado de cancro da mama e/ou ovário, são:
(* dados limitados)Probabilidade de Cancro ao longo da vida Cancro da Mama Cancro do Ovário Cancro do Pâncreas ou outros cancros PALB2 41-61% 3-5% 3-5% ATM 15-40% <3% 5-10% CHEK2 15-40% Sem evidência Cólon CDH1 41-60% Sem evidência Cancro Gástrico Difuso RAD51C 15-40%* >10% Evidência insuficiente RAD51D 15-40%* >10% Evidência insuficiente
Como é herdado o HBOC?O risco de transmissão hereditária da cópia do gene alterada de um progenitor para os seus filhos é de 50% - transmissão autossómica dominante, independentemente se é o pai ou a mãe, o progenitor afetado.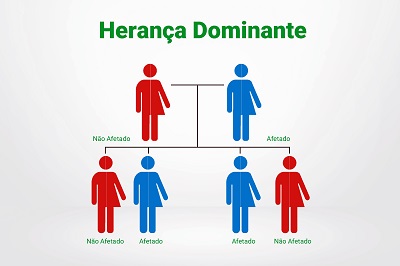
Quais as opções de vigilância?A vigilância depende do nível de risco de vir a ter cancro, o qual varia de acordo com o gene alterado e com a história familiar de cancro.Portadoras de variantes causais (patogénicas ou provavelmente patogénicas) dos genes BRCA1 e BRCA2 Protocolo de vigilância: Mulheres com Síndrome Hereditário de cancro da mama/ovário
Protocolo de vigilância: Mulheres com Síndrome Hereditário de cancro da mama/ovário
Descarregue o guia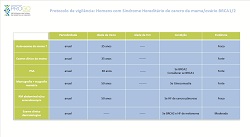
Protocolo de vigilância: Homens com Síndrome Hereditário de cancro da mama/ovário
Descarregue o guia
A ecografia mamária em conjugação com a mamografia pode ser considerada em todas as idades como alternativa à RM mamária se esta não estiver disponível (IV,B)Não existe atualmente evidência que a ecografia ginecológica endovaginal e CA125 semestral/anual, contribuam para o aumento da deteção precoce, reduzam a mortalidade ou aumentem a sobrevivência associada ao cancro do ovário em mulheres com alto risco.Nas mulheres submetidas a histerectomia (BRCA1) podem ser candidatas a utilizar terapêutica hormonal com estrogénio isolado.Quais as opções redutoras de risco de cancro?1. Medidas gerais- Manter uma alimentação equilibrada - pobre em gorduras, sal e acuçar e abandonate em alimentos frescos, ricos em vitaminas, fibra e antioxidantes. A carne vermelha e carne processada devem ser evitadas.- Realizar exercício físico regular (atividade aerobica moderada >= 50 min/semana ou vigurosa >= a 75min/semana e >= 2 treinos de resitência por semana).- Manter peso saudável (indice de Massa Corporal de 18,5 a 24,9)- Limitar consumo de álcool
- Proteger-se do sol- Ter filhos e amamentar2. QuimioprevençãoOs resultados da quimioprevenção em mulheres BRCA1/2 são controversos, pelo que as cirurgias redutoras de risco são a estratégia mais eficaz.A utilização dos moduladores seletivos dos recetores de estrogénio (tamoxifeno ou raloxifeno) parece diminuir o risco de cancro da mama invasivo nas portadoras de variantes causais BRCA1 e BRCA2 na pós-menopausa. Contudo, os dados disponíveis da sua utilização são muito limitados e não se encontram aprovados, neste contexto, em Portugal.Também anastrozole e exemestano demonstraram redução do risco relativo de incidência de cancro invasivo em mulheres pós-menopausicas com risco aumentado de cancro da mama.Nas mulheres com diagnóstico prévio de cancro da mama está demonstrado o benefício do tratamento hormonal adjuvante como estratégia de redução de risco de cancro da mama contralateralNas mulheres portadoras de variantes patogénicas do gene BRCA1/BRCA2 sem história de neoplasia da mama e já submetidas a cirurgia mamária redutora de risco poderá ser ponderado o uso de contracetivos orais como estratégia redutora de risco para cancro do ovário.3. Cirurgias redutoras de risco
Mastectomia subcutânea bilateral com preservação do complexo areolo-mamilar – reduz o risco de cancro da mama em mais de 90%. Só cerca de 3% do cancro da mama associado a mutações BRCA1/2 são diagnosticados antes dos 30 anos, pelo que este tipo de cirurgia poderá ser equacionado após esta idade.Salpingo-ooforectomia bilateral (cirurgia de remoção dos ovários e trompas de fallopio) claramente a melhor estratégia redutora de risco de cancro do ovário. Tem sido também utilizada como estratégia redutora de risco de cancro da mama, contudo resultados de estudos mais recentes revelam que esta proteção diminui após os 5 anos da cirurgia.A decisão deverá ter em consideração o desejo reprodutivo e apenas deverá ser efetuada após estar completo o projeto reprodutivo:- Mulheres portadoras de BRCA1 - aos 35-40 anos (eventual histerectomia)- Mulheres portadoras de BRCA2 – aos 40-45 anosEsta cirurgia está associada à menopausa cirúrgica com efeitos secundários muito marcados sobretudo nas mulheres jovens. Nas situações onde não existem antecedentes pessoais de cancro da mama, a evidencia científica sugere que a terapêutica hormonal de substituição não aumenta o risco de cancro da mama, podendo ser utilizada para controlo sintomática e melhoria da qualidade de vida. Poderá ser instituída pelo menos até à idade da menopausa natural (50-51 anos). Nas mulheres com antecedentes de cancro da mama e menopausa cirúrgica o tratamento hormonal de substituição está contraindicado.Que vigilância após cirurgias redutoras de risco:1. Vigilância mamária após mastectomia redutora de riscoApós a cirurgia deverá ser efetuada RM mamária para avaliação e quantificação do tecido mamário remanescente.- Se abundante tecido mamário remanescente - a vigilância deverá ser mantida com RM mamária anual.- Se inexistente ou muito reduzido – a vigilância deverá ser feita apenas com exame clínico anual. Neste caso a avaliação imagiológica deve ser decidida de acordo com a observação clínica.2. Vigilância ginecológica após salpingoooforectomia- Deve ser mantida vigilância ginecológica regular (caso mantenha útero), sobretudo para rastreio de patologia uterina.
- Deve ser mantido o doseamento do CA125 anual.Referências bibliográficasNCCN Clinical Practice Guidelines in Oncology - Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic (version 2.2021 - November 20, 2020)Prevention and screening in BRCA mutation carriers and other breast/ovarian hereditary cancer syndromes: ESMO Clinical Practice Guidelines for cancer prevention and screening, S. Paluch-Shimon et al on behalf of the ESMO Guidelines Committee, Annals of Oncology 27 (Supplement 5): v103–v110, 2016.
Brennan A. et all. Int J Gynecol Cancer 2021; 31: 352-359Yun-Hee Choi, PhD; Mary Beth Terry, PhD; Mary B. Daly,MD, PhD et all. Association of Risk-Reducing Salpingo-Oophorectomy With Breast Cancer Risk inWomen With BRCA1 and BRCA2 Pathogenic Variants Int JAMA Oncology 2021; 7: 585-892
- Cancro Gástrico Difuso Hereditário
-
O que é a cancro gástrico difuso hereditário (HDGC)?O HDGC é uma síndrome associada ao desenvolvimento de cancro, caracterizada pelo desenvolvimento de cancro de estômago e da mama, preferencialmente antes dos 50 anos. Os tipos de cancro de estômago e mama associados a esta síndrome são específicos e são eles: cancro de estômago de tipo difuso e cancro da mama de tipo lobular.O cancro de estômago é o tipo de cancro mais frequente nas famílias com HDGC, seguido pelo cancro da mama. Gerações sucessivas são frequentemente afetadas por esses tipos de cancro que surgem geralmente a partir dos 20 ou 30 anos de idade.O que causa HDGC?O HDGC é causado por alterações genéticas, geralmente herdadas em famílias. As alterações mais frequentes ocorrem numa das cópias (herdadas da mãe ou do pai) do gene CDH1 que codifica a proteína Caderina-E, e afetam entre 10-40% das famílias que apresentam critérios clínicos desta doença.Recentemente, algumas famílias com HDGC revelaram a presença de alterações genéticas no gene CTNNA1 que codifica a proteína a-E-catenina, e que explicam menos de 2% dos casos.Estas duas proteínas têm em comum a função de manutenção da adesão entre células epiteliais e a prevenção da migração e invasão destas células para outros tecidos. Quando estas funções são perturbadas por alterações genéticas, os cancros desenvolvem-se, ganham mobilidade e tornam-se muito agressivos.Como é transmitida a síndrome HDGC?O HDGC é uma síndrome autossómica dominante, o que significa que todas as gerações familiares tendem a ter indivíduos afetados com cancro de estômago ou da mama. Indivíduos que herdam uma única cópia do gene mutante (CDH1 ou CTNNA1) têm maior risco de vir a desenvolver cancro de estômago, independentemente de serem homens ou mulheres. Por outro lado, apenas mulheres portadoras de alterações no gene CDH1 têm risco de desenvolver cancro da mama. Não há conhecimento atualmente de que alterações no gene CTNNA1 predisponham para cancro da mama. O HDGC é transmitido por um progenitor (pai ou mãe) portador de uma alteração nos genes CDH1 ou CTNNA1. Os filhos têm 50% de probabilidade de receber a alteração genética.Como se manifesta?Os portadores masculinos de alterações patogénicas no gene CDH1 têm um risco aumentado de poderem desenvolver cancro do estômago ao longo da sua vida. As mulheres portadoras têm um risco aumentado de desenvolver tanto cancro do estômago como cancro da mama ao longo da vida. Dependendo da variante patogénica, e da população estudada:• O risco de cancro de estômago à idade de 80 anos, é de 42% em homens e de 33% em mulheres, se ambos forem portadores de uma variante patogénica no gene CDH1.• As mulheres portadoras de uma variante patogénica no gene CDH1 têm um risco adicional, que pode chegar a 55% à idade de 80 anos, de desenvolver cancro da mama.Não há atualmente cálculos de risco para os portadores de variantes patogénicas no CTNNA1.Contudo, nem todas as pessoas com variantes patogénicas germinativas nos genes CDH1 e CTNNA1 vêm a desenvolver cancro.
Probabilidade de cancro ao longo da vida Cancro do estômago em homens Cancro do estômago em mulheres Cancro da mama e estômago em mulheres População
Geral<2% <1% Não disponível CDH1 42-70% 33-56% 39-55% CTNNA1 Não disponível Não disponível Não disponível Quais são as opções de vigilância para HDGC?A opção preventiva primária da síndrome HDGC é a remoção do estômago (gastrectomia total) em pessoas que são portadoras de variantes patogénicas do gene CDH1, e antes da manifestação da doença. Caso a gastrectomia não seja apropriada por razões clínicas, ou recusada pelo portador da variante genética, a vigilância pode ser efetuada com gastroscopia anual e biópsias múltiplas.Em mulheres portadoras de variantes patogénicas do gene CDH1, a mamografia anual e a ressonância magnética das mamas, a partir dos 35 anos, é o procedimento de vigilância mais adequado para a deteção precoce de cancro da mama lobular. A mastectomia é oferecida a doentes com lesões precoces de cancro da mama e geralmente é curativa. A remoção preventiva de ambas as mamas (mastectomia bilateral) também é uma opção, mas deve ser cuidadosamente discutida com a equipa clínica.Em portadores de variantes patogénicas do gene CTNNA1, a vigilância de cancro do estômago deve ser efetuada com gastroscopia anual e biópsias múltiplas, devendo a remoção profilática do estômago (gastrectomia total) ser discutida em consulta multidisciplinar e o risco calculado com base na história familiar.Em mulheres portadoras de variantes patogénicas do gene CTNNA1, a vigilância das mamas e eventual mastectomia profilática, deve apenas ser considerada se houver história familiar de cancro lobular da mama em portadoras na família, e deve ser discutida com os portadores e doentes.Os portadores de alterações nos genes CDH1 ou CTNNA1 devem ser acompanhados por uma equipa multidisciplinar de especialistas nesta doença e com experiência reconhecida no seu diagnóstico e tratamento

Protocolo de vigilância de cancro de estômago em portadores de variantes no gene CDH1 e com critériosclínicos de síndrome de HDGC
 Protocolo de vigilância de cancro da mama em portadores de variantes no gene CDH1 e com critérios clínicosde síndrome de HDGCDescarregue o guiaPara mais informações consultar https://www.sciencedirect.com/science/article/pii/S1470204520302199?via%3Dihub
Protocolo de vigilância de cancro da mama em portadores de variantes no gene CDH1 e com critérios clínicosde síndrome de HDGCDescarregue o guiaPara mais informações consultar https://www.sciencedirect.com/science/article/pii/S1470204520302199?via%3Dihub
Bibliografia:
Blair VR, McLeod M, Carneiro F, Coit DG, D'Addario JL, van Dieren JM, Harris KL, Hoogerbrugge N, Oliveira C, et al. Hereditary diffuse gastric cancer: updated clinical practice guidelines. Lancet Oncol. 2020. doi: 10.1016/S1470-2045(20)30219-9.
- Síndrome Li-Fraumeni
-
O que é a síndrome de Li-Fraumeni (SLF)?A síndrome de Li-Fraumeni é uma doença rara que aumenta muito o risco de desenvolver vários tipos de cancro, principalmente em crianças e adultos jovens.O diagnóstico deve ser considerado se um doente apresentar uma das seguintes situações clínicas:1. Doente com:a. Idade inferior a 46 anos e tumor sugestivo da síndrome SLF (tumor de tecidos moles – sarcoma; tumor ósseo; tumor na camada externa das glândulas adrenais - carcinoma adrenocortical; tumor cerebral, cancro da mama em mulheres), e que;a.1. Apresente pelo menos um parente de primeiro ou segundo grau com um tumor do espetro SLF antes dos 56 anos de idade, OU;a.2. Apresente pelo menos um parente de primeiro ou segundo grau com tumores múltiplos.2. Doente com:a. Dois tumores pertencentes ao espectro da síndrome SLF (ver em a.), tendo-se manifestado o primeiro antes dos 46 anos, OU;b. Um segundo tumor desenvolvido em campo irradiado durante o tratamento de um primeiro tumor.3. Doente com um tumor muito raro, como um tumor adrenocortical, um tumor cerebral raro denominado tumor dos plexos coroideus, ou um tipo específico de sarcoma denominado rabdomiossarcoma anaplásico embrionário.4. Doente do sexo feminino com cancro da mama antes dos 31 anos.O que causa síndrome de Li-Fraumeni?A síndrome de Li-Fraumeni resulta da presença de uma alteração no gene TP53 que codifica a proteína p53. Estas alterações podem ser herdadas de um dos progenitores; ocorrer nas células sexuais dos pais (espermatozoides ou óvulos); ou ocorrer durante o desenvolvimento embrionário. A proteína p53, quando na sua forma normal, atua como guardiã do genoma e previne a ocorrência de danos no DNA.Nos doentes que apresentam uma alteração no gene TP53, a quantidade da proteína normal é insuficiente e, quando ocorrem danos ao DNA numa célula, estes não são corrigidos. A acumulação de erros sucessivos e sem possibilidade de correção, levam a que células normais se tornem cancerígenas.Como ocorre e como é transmitida?A síndrome de Li-Fraumeni pode ocorrer de três formas:1- Ser transmitida de forma autossómica dominante, ou seja, um progenitor (pai ou mãe), quando portador de uma alteração no gene TP53 tem 50% de probabilidade de a transmitir aos filhos. Na maioria das famílias, a alteração genética é herdada da mãe ou do pai.2- Ocorrer de novo, ou seja, está apenas presente nos espermatozoides ou óvulo dos pais, podendo originar uma criança em que todas as células apresentam a alteração e por isso com risco para síndrome de Li-Fraumeni. Estes casos ocorrem sem que os pais sejam portadores da alteração genética e ocorre em pelo menos 14% das situações.3- Ocorrer por mosaicismo somático clássico, ou seja, ocorre uma alteração no gene TP53 durante as primeiras fases do desenvolvimento embrionário e apenas em algumas células do embrião. Estas alterações podem ou não causar doença dependendo do tecido em que estão presentes após o nascimento. Portanto, os indivíduos-mosaico expressarão doença dependendo do número e tipo de células afetadas pela alteração genética. Atualmente, desconhece-se a frequência desta situação.O risco de um portador de uma alteração no gene TP53 desenvolver cancro é bastante diverso, mesmo dentro da mesma família. Uma família portadora pode então apresentar muitos cancros ou muito poucos. O risco depende do tipo de alteração e provavelmente da presença de outras variações genéticas que atuam como fatores promotores ou protetores.A radioterapia e as quimioterapias genotóxicas contribuem para o desenvolvimento de novos tumores em portadores de alterações do TP53, a maioria nas zonas sujeitas a irradiação. Portanto, a radioterapia deve ser evitada e o tratamento cirúrgico priorizado, mas somente se isso for possível e não comprometer o tratamento do cancro.
Como se manifesta?Os portadores de alterações patogénicas no gene TP53 têm um risco aumentado de virem a desenvolver vários cancros ao longo da sua vida, e a partir do nascimento.Os cálculos de risco para cancro nos portadores de alterações do TP53 foi durante muito tempo sobrestimado, dado que foram analisados preferencialmente famílias com variantes herdadas e que apresentavam critérios clínicos de síndrome de Li-Fraumeni. A incidência cumulativa de cancro em portadores de variantes TP53 causadoras de doença foi portanto inicialmente estimada em 73-100% aos 70 anos, com riscos próximos a 100% em mulheres.Atualmente, sabe-se que há muitos portadores de alterações do TP53 sem história familiar, que vêm a desenvolver cancro, o que torna o estudo do risco para cancro um assunto complexo e desconhecido até ao momento.Estima-se que a prevalência de alterações germinativas do TP53 na população geral seja 1 por cada 4.500 indivíduos. É importante salientar que nem todas as pessoas portadoras de alterações patogénicas no gene TP53 vêm a ter cancro.Na infância, os principais riscos de cancro em portadores de alteração de TP53 são carcinomas adrenocorticais, tumores de tecidos moles ou sarcomas, osteossarcomas, tumores cerebrais e do sistema nervoso central e tumores na camada externa das glândulas adrenais ou carcinomas adrenocorticais.O principal risco de cancro em adultos corresponde a cancro da mama em mulheres portadoras de alterações no TP53. Estas apresentam um risco muito alto para desenvolver cancro da mama antes dos 31 anos de idade. Não há risco reconhecido de cancro da mama no homem em portadores de alteração no TP53.Um fator que explica a variabilidade da penetrância de doença em portadores de alterações no TP53 é o tipo da própria variante: algumas das proteínas p53 com mutações missense são classificadas como tendo efeito dominante-negativo devido à sua capacidade de reduzir a atividade da proteína p53. Esse tipo de variantes no TP53 são geralmente detectadas em famílias com cancros na infância e geralmente são altamente penetrantes.Ao contrário, as variantes nulas (frameshift, nonsense, de splicing, grandes rearranjos genómicos e variantes missense sem efeito dominante-negativo) são predominantemente identificadas em famílias com cancros na idade adulta e apresentam baixa penetrância.Um exemplo notável de uma variante e baixa penetrância, mas ainda assim causadora de doença, é a variante missense sem efeito dominante-negativo p.Arg337His, presente em 0,3% da população do Sul do Brasil e associada a um efeito fundador. Algumas famílias portuguesas também apresentam esta variante.
Quais são as opções de vigilância para a síndrome de Li-Fraumeni?Os protocolos de vigilância podem ser iniciados no primeiro ano de vida, e são baseados em ultrassonografia abdominal a cada 3-6 meses, ressonância magnética corporal total anual, ressonância magnética cerebral anual e, em mulheres a partir dos 20 anos, em ressonância magnética de mama anual. Os benefícios destes protocolos pesados devem ser cuidadosamente analisados e discutidos em cada família e em centros com experiência demonstrada. Protocolo de Vigilância para Síndromes de Li-Fraumeni e cancro relacionado com a TP53 hereditário (hTP53rc)Descarregue o guiaReferência Bibliográfica:Frebourg, T., Bajalica Lagercrantz, S., Oliveira, C. et al. Guidelines for the Li–Fraumeni and heritable TP53-related cancer syndromes. Eur J Hum Genet 28, 1379–1386 (2020). https://doi.org/10.1038/s41431-020-0638-4
Protocolo de Vigilância para Síndromes de Li-Fraumeni e cancro relacionado com a TP53 hereditário (hTP53rc)Descarregue o guiaReferência Bibliográfica:Frebourg, T., Bajalica Lagercrantz, S., Oliveira, C. et al. Guidelines for the Li–Fraumeni and heritable TP53-related cancer syndromes. Eur J Hum Genet 28, 1379–1386 (2020). https://doi.org/10.1038/s41431-020-0638-4